INFORMATION ÜBER DIE
SICHELZELLKRANKHEIT
Was ist die Sichelzellkrankheit?
Es ist eine weltweit verbreitete erbliche Erkrankung der roten Blutkörperchen, die sich aber im ganzen Körper und in fast allen Organen auswirken kann.
Warum sind bei der Sichelzellkrankheit die roten Blutkörperchen krank?
Rote Blutkörperchen, die man auch Erythrozyten nennt, sind kleine, runde Scheiben, die im Blut den Sauerstoff von der Lunge in alle Teile des Körpers bringen. Der Sauerstoff ist in den roten Blutkörperchen an das Hämoglobin gebunden. Das Hämoglobin heisst auch roter Blutfarbstoff, da durch ihn das Blut die rote Farbe hat.
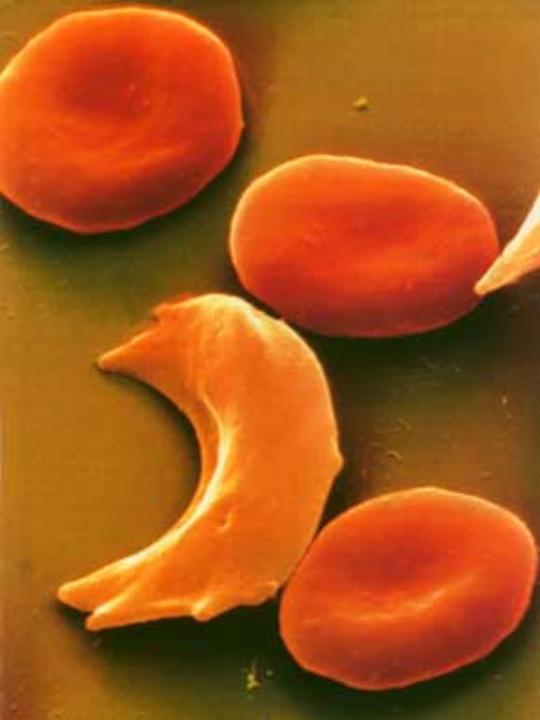
Wenn man die Sichelzellkrankheit hat, hat man im Blut nicht den normalen roten Blutfarbstoff, den man Hämoglobin A (HbA) nennt, sondern einen krankhaft veränderten roten Blutfarbstoff, das Hämoglobin S (HbS). Dieser veränderte rote Blutfarbstoff macht aus den runden, glatten, weichen und gut im Blutstrom beweglichen roten Blutkörperchen spitze, harte, lange Zellen, die wie Sicheln aussehen und die deshalb den Namen Sichelzellen bekommen haben.
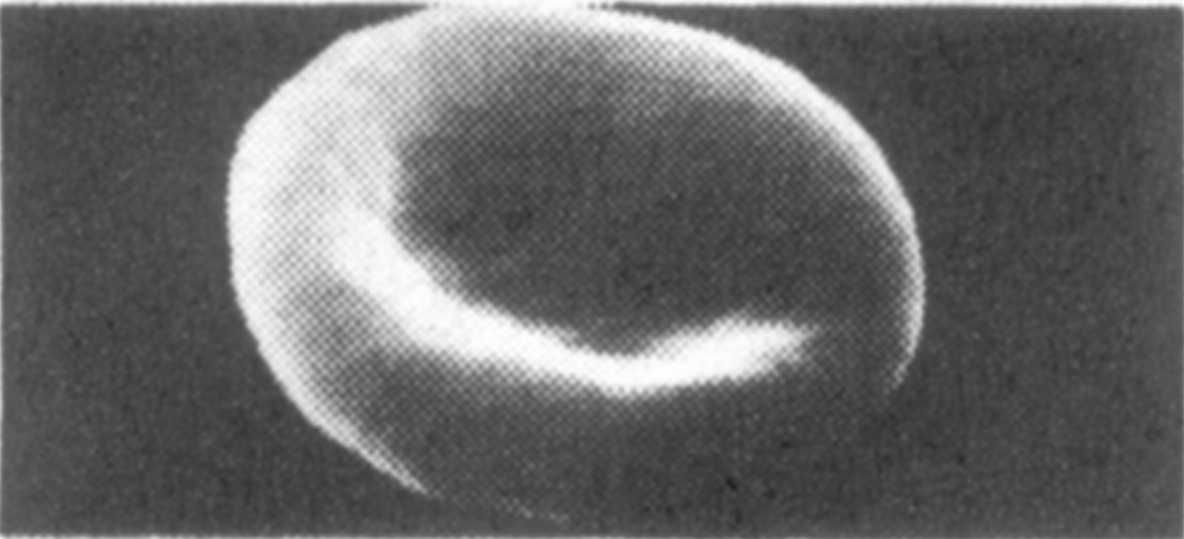
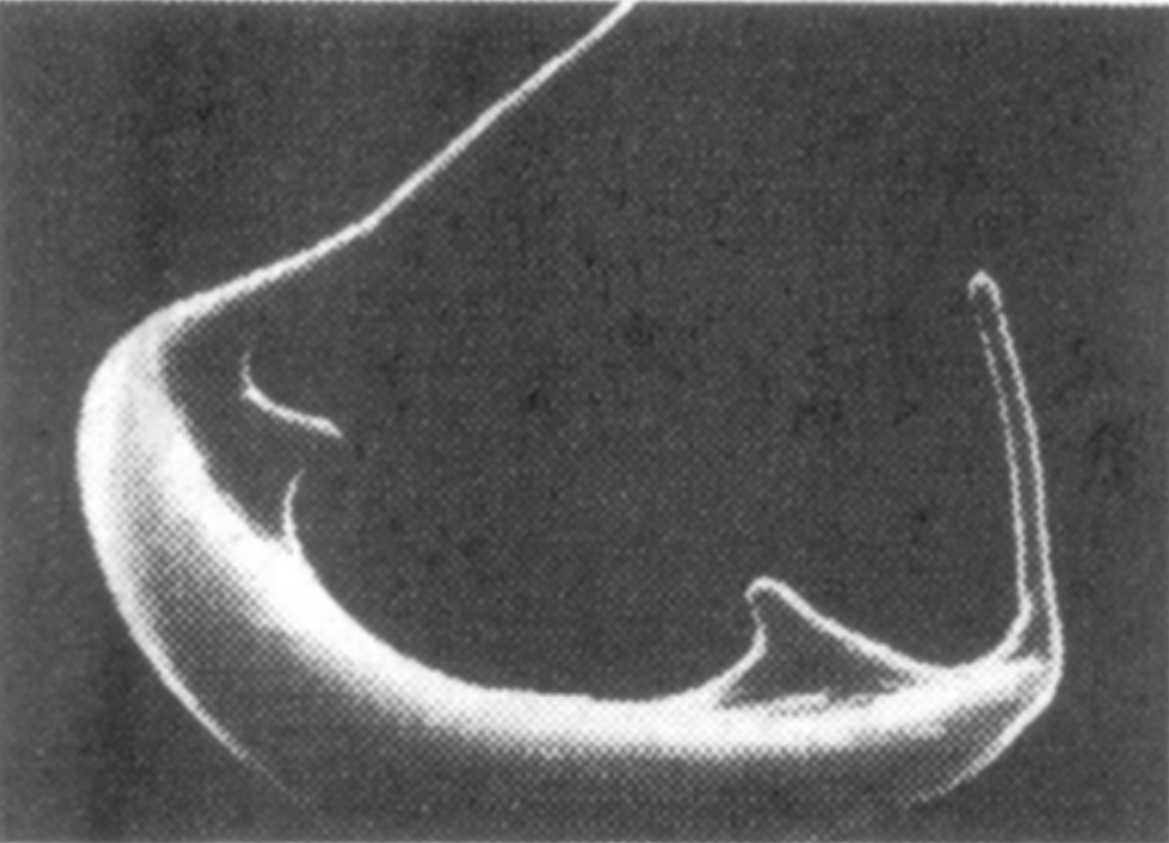
Abb. 1
links (oben) : Gesundes rotes
Blutkörperchen
Abb. 1 rechts
(unten): Sichelzelle
Wo gibt es die Sichelzellkrankheit?
Die Veränderung der
Erbanlage (=Mutation) für das Hämoglobin S ist vor vielen
hundert Jahren in West-und Zentralafrika und auf der
Arabischen Halbinsel entstanden. Von diesen Gegenden
aus hat sich das veränderte Gen und die Krankheit nach allen
Richtungen ausgebreitet. Wir finden Sichelzellkrankheiten
heute also auch im östlichen Mittelmeerraum
(Griechenland, Süditalien, Türkei), im Mittleren Osten
(Libanon, Syrien, Irak, Arab. Halbinsel), Indien, Pakistan.
Nach Nord-und Südamerika ist die Erkrankung durch den
Sklavenhandel gelangt.
In der Türkei
kommt die Sichelzellkrankheit hauptsächlich im Süd-Osten vor,
in der Gegend um Adana, Mersin, Iskenderun. In Nordeuropa, d.
h. in Deutschland, Frankreich, England, Holland, Belgien,
Skandinavien gab es die Erkrankung früher nicht. Sie ist
durch Einwanderer aus dem Mittelmeerraum, Afrika und Asien zu
uns gekommen.
Warum wird man krank, wenn man Sichelzellen hat?
1. Sichelzellen
können in den Blutgefäßen steckenbleiben, weil sie nicht
so glatt, rund und beweglich sind wie die gesunden roten
Blutkörperchen. Wenn ein
Gefäß durch
Sichelzellen verstopft ist, kommt kein Sauerstoff mehr
in das Gewebe. Sauerstoffmangel führt zur Zerstörung des
Gewebes und es werden Stoffe gebildet, die Schmerzen auslösen.
Heftige Schmerzen, die auch als Schmerzkrisen bezeichnet
werden, entstehen vor allem in den Knochen durch Verschluß von
Blutgefäßen im Knochenmark. Werden Blutgefäße in anderen
Organen (Lunge, Niere, Gehirn, Leber) durch Sichelzellen
verschlossen, kann es zu Schädigung dieser Organe kommen.
2. Sichelzellen zerfallen schneller als gesunde, runde Blutkörperchen und es entsteht eine Blutarmut (=Anämie), obwohl das Knochenmark vermehrt arbeitet. Da fast alle Sichelzellpatienten eine chronische Anämie haben, wurde die Sichelzellkrankheit früher auch Sichelzellanämie genannt. Dieser Name sollte nicht mehr verwendet werden da er nicht korrekt ist:es handelt sich um eine Krankheit, die alle Organe betreffen kann.
3. Sichelzellen verstopfen besonders häufig, meist schon im ersten Lebensjahr, die Gefäße der Milz. Die Milz ist ein sehr wichtiges Organ zur Abwehr einiger Bakterien (z. B. Pneumokokken). Wenn immer wieder Gefäße verschlossen werden, wird das Milzgewebe so geschädigt, dass es seine Aufgabe, den Körper vor Pneumokokken zu schützen,nicht mehr erfüllen kann. Sichelzellpatienten, vor allem Kinder in den ersten Lebensjahren, sind gefährdet durch Infektionen, vor allem aber Pneumokokken-Infektionen, die lebensbedrohlich sind, wenn sie nicht rechtzeitig erkannt werden. Um Kinder mit Sichelzellkrankheiten vor diesen Infektionen zu schützen, müssen sie vom 3. Lebensmonat bis zum 5. Lebensjahr täglich Penizillin nehmen. Es gibt auch eine Impfung gegen Pneumokokken, die ab dem 3. Lebensmonat gegeben werden soll.
Wie bekommt man die Sichelzellkrankheit?
Die
Sichelzellkrankheit wird vererbt. Jeder Mensch hat für
alle Eigenschaften seines Körpers, wie Haarfarbe, Größe, Farbe
der Augen usw. zwei Erbanlagen (=Gene), eine vom Vater und
eine von der Mutter. Auch die Zusammensetzung unseres roten
Blutfarbstoffes wird von Genen bestimmt, die sowohl vom Vater
als auch von der Mutter stammen. Wenn man vom Vater und von
der Mutter die Gene für den normalen roten Blutfarbstoff, das
Hämoglobin A, geerbt hat, hat man gesunde rote Blutkörperchen.
Wenn man vom Vater und der Mutter die Gene für den
veränderten Blutfarbstoff, das Hämoglobin S, geerbt hat, hat
man die Sichelzellkrankheit HbSS.
Im Blut ist nur verändertes Hämoglobin, das die roten
Blutkörperchen lang und spitz, also sichelförmig werden läßt.
Es gibt ausser dem
Gen für das Hämoglobin S noch andere Gene, die den roten
Blutfarbstoff krankhaft verändern. Wenn jemand von einem
Elternteil das Hämoglobin S geerbt hat und vom anderen das Gen
für entweder die ß-Thalassämie oder das krankhafte Hämoglobin
C, D, OArab oder Lepore, hat er ebenfalls eine
Sichelzellkrankheit: HbSßThal, HbSC, HbSD, HbSOArab,
HbSLepore.
Hat man eine Sichelzellkrankheit, wenn man nur von einem Elternteil das Gen für das Hämoglobin S erbt, vom anderen aber das normale Hämoglobin A?
Nein. Wenn jemand nur von einem Elternteil das Gen für das veränderte Hämoglobin S erbt, vom anderen Elternteil aber das Gen für den normalen roten Blutfarbstoff, Hämoglobin A, dann ist dieser Mensch ein Träger der Sichelzell-Erbanlage. Ein Träger hat ca. 40% Hämoglobin S in seinem Blut, ist nicht krank, kann allerdings die Erbanlage für die Erkrankung an seine Kinder weitergeben. Deshalb ist es wichtig zu wissen, ob man Träger ist. Die Trägerschaft verändert das Blutbild nicht. Um herauszufinden, ob jemand Träger der Erbanlage für das Hämoglobin S ist, muß eine Blutuntersuchung, eine Hämoglobin-Analyse, in einem Spezial-Labor gemacht werden.
Wie stellt man fest daß jemand die Sichelzellkrankheit hat?
Wenn jemand aus einem der Länder kommt, in denen die Sichelzellkrankheit vorkommt, und eine Blutarmut (=Anämie) hat und/oder häufige, unklare Knochenschmerzen, muß eine Hämoglobin-Analyse durchgeführt werden, um die Diagnose zu stellen. In USA, Frankreich, England, Belgien, Holland wird jedes Neugeborene, dessen Eltern aus Ländern kommen, in denen es die Sichelzellkrankheit gibt, untersucht, ob es die Krankheit hat. In Deutschland gibt es seit Oktober 2021 das allgemeine Neugeborenen-Screening. Jedes Neugeborene, unabhängig von der Herkunft der Eltern, wird auf die Sichelzellkrankheit untersucht.
Warum habe ich die Sichelzellkrankheit und meine Schwester (mein Bruder) nicht?
Wie bereits erwähnt
hat jeder Mensch doppelte Erbanlagen (Gene) für alle
Körpereigenschaften, auch die des Blutes, von seinen Eltern
geerbt. Wenn ein Kind geboren wird, hat es ein Gen vom Vater,
das andere von der Mutter in sich.
Wenn jemand Träger
der Sichelzellanlage ist, hat er zwei unterschiedliche Gene
für den roten Blutfarbstoff : ein Gen für das gesunde, normale
Hämoglobin, das Hämoglobin A, und ein verändertes Gen für das
Hämoglobin S. Ein Träger gibt entweder das gesunde Gen an sein
Kind weiter, oder das veränderte. Wenn beide Eltern Träger des
Gens für Hämoglobin S sind, gibt es drei Möglichkeiten für die
Kombination von Genen, die die Kinder erben können. Welches
Gen das Kind vom Vater oder der Mutter erbt ist rein
zufällig.
1. Das Kind erbt von
beiden Eltern deren gesundes, normales Gen für Hämoglobin: das
Kind ist gesund und hat nur Hämoglobin A.
2. Das Kind erbt von
beiden Eltern deren verändertes Gen, das Gen für Hämoglobin
S. Das Kind hat die Sichelzellkrankheit und hat kein
normales Hämoglobin A.
3. Das Kind erbt von
einem Elternteil das gesunde, vom anderen das veränderte Gen.
Das Kind ist gesund, hat ca. 35-40% Hämoglobin S, aber auch
> 50% normales Hämoglobin A in seinem Blut. Es trägt
die Anlage für die Sichelzellkrankheit und kann sie an seine
eigenen Kinder weitergeben.
Abb2. Beide Partner tragen die HbS-Anlage (AS): es können kranke (SS) und gesunde Kinder (AA, AS) geboren werden.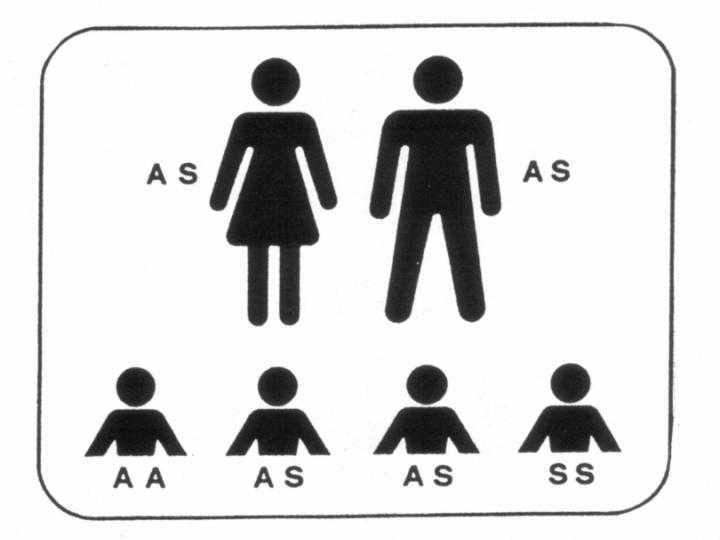
Kann man die Sichelzellkrankheit heilen?
Ja. Allerdings nicht
durch Medikamente, sondern nur durch eine Knochenmark
(Stammzell) - Transplantation. Da eine
Knochenmarktransplantation mit Komplikationen verbunden ist
und in einigen Fällen auch tödlich ausgeht, ist es eine
Therapieform, die bis jetzt für nur wenige Sichelzellpatienten
gewählt wurde. Weltweit wurden bis 2017 ca. 1000
Sichelzellpatienten transplantiert, fast alle waren Kinder
< 16 Jahre. Allen Patienten mit der Form HbSS, HbSß°Thal,
HbSD, HbSOArab, die einen HLA-identischen Familienspender
haben, sollte die Transplantation angeboten werden. Inzwischen
ist ein Verfahren (Konditionierung) entwickelt worden, das es
erlaubt, auch Erwachsene mit Sichelzellkrankheit zu
transplantieren, allerdings nur, wenn es einen passenden
(HLA-identischen) Spender gibt. Die sog. haploidentische
Transplantation von einem Spender (Eltern oder Geschwister),
der nur zu 50% HLA-identisch mit dem Patienten ist, muß noch
als experimentell betrachtet werden und sollte nur im Rahmen
von Studien durchgeführt werden.
Seit Oktober 2019 wird in einer Studie eine Gen-Therapie
angeboten, mit der es allerdings noch sehr wenig Erfahrung
gibt.
Kann man die Sichelzellkrankheit behandeln?
Ja. Kinder unter 5
Jahren können vor lebensbedrohlichen Pneumokokken-Infektionen
geschützt werden durch die tägliche Gabe von Penizillin
und durch eine Impfung gegen Pneumokokken. Infektionen
werden mit einem der Infektion entsprechenden Antibiotikum
behandelt. Bei Schmerzkrisen müssen neben ausreichender
Flüssigkeit Schmerzmittel gegeben werden, die der Schwere der
Schmerzen entsprechen, d. h. manchmal sind starke Mittel wie
Morphium notwendig für einige Tage.
Bei einigen
Komplikationen der Erkrankung müssen Bluttransfusionen
gegeben werden.
Seit einigen Jahren
wissen wir, dass Sichelzellpatienten, die sehr häufige und
schwere Schmerzkrisen bzw. häufige schwere
Lungenkomplikationen haben, wesentlich weniger Probleme haben
wenn sie Hydroxycarbamid nehmen. Hydroxycarbamid ist
ein Medikament, das zu den sog. Zytostatika gehört, d. h. zu
den Medikamenten, die man u. a. auch bei Leukämien einsetzt.
Hydroxycarbamid verändert nicht nur die Zusammensetzung des
roten Blutfarbstoffes, es beeinflusst auch die roten
Blutkörperchen und hilft mit, dass sie die Blutgefässe nicht
verstopfen. Es gibt inzwischen Erfahrung mit der
langfristigen Gabe dieses Medikamentes. Bei Patienten, die 20
Jahre lang Hydroxycarbamid genommen haben, sind nicht mehr
bösartige Krankheiten aufgetreten als bei Patienten, die es
nicht nahmen. Es scheint auch keine Fehlbildungen bei Kindern
zu verursachen, deren Mütter oder Väter zur Zeit der Zeugung
Hydroxycarbamid genommen haben. Seit 2014 wird empfohlen,
allen Sichelzellpatienten mit den Formen HbSS, HbSD,
HbSß°Thal, HbSOArab ab dem 9. Lebensmonat
Hydroxycarbamid zu geben, da es nicht nur Schmerzkrisen und
Akute Thorax-Syndrome, sondern auch Schäden an anderen
Organen, z. B. der Nieren, verhindern kann.
Bei einigen
Komplikationen (z. B. Gallensteine, die Beschwerden
verursachen; sehr große Milz, die die Blutzellen vernichtet;
Schmerzen in den Hüftgelenken) kann durch eine Operation
geholfen werden.
Kann man etwas tun um Schmerzkrisen zu verhindern?
Ja. Da Schmerzkrisen
ausgelöst werden können durch Unterkühlung, Rauchen, Alkohol,
zu wenig Flüssigkeit und Infektionen sollten
Sichelzellpatienten sich folgendermaßen verhalten:
Nicht rauchen!
Wenig, wenn überhaupt, Alkohol! Nicht in kaltem Wasser
schwimmen; sich bei Kälte entsprechend warm anziehen;
Sichelzellpatienten
müssen immer mehr trinken als die anderen, da ihre Nieren den
Urin nicht konzentrieren können: vor allem bei warmem Wetter,
Sport, körperlichen Anstrengungen; bei Flugreisen müssen
Sichelzellpatienten zusätzlich große Mengen trinken, da die
Luft im Flugzeug sehr trocken ist.
Bei Fieber >
38,5°C muß der Arzt aufgesucht werden, damit festgestellt
wird, ob es sich um eine Infektion handelt, bei der
Antibiotika notwendig sind.
Bei ca. 70% der
Patienten mit schweren bzw. sehr häufigen Schmerzkrisen können
diese verhindert oder verringert werden durch Hydroxycarbamid.
Dürfen Sichelzellpatienten Sport treiben?
Ja. Die meisten Sichelzellpatienten können und sollen Sport treiben, allerdings keinen Leistungssport. Durch die Blutarmut werden Sichelzellpatienten schneller müde bei körperlicher Anstrengung. Deshalb muß mit den Sportlehrern abgesprochen werden, dass Kinder und Jugendliche mit Sichelzellkrankheit selber bestimmen dürfen, wo ihre Grenzen sind.
Kann man die Sichelzellkrankheit verhindern?
Ja. Wenn Vater und Mutter Träger des Gens für die Sichelzellkrankheit sind, gibt es die Möglichkeit der Pränatal-Diagnose. Früh in der Schwangerschaft, zwischen der 10.-12. Woche wird ein kleines Stück Gewebe aus der Plazenta entnommen, das Zellen des Kindes enthält. Untersuchungen dieser kindlichen Zellen können zeigen, ob das Kind gesund ist, d. h. entweder nur gesunde Gene für den roten Blutfarbstoff hat oder gesunder Träger oder ob es die Sichelzellkrankheit geerbt hat. In diesem Fall kann die Schwangerschaft abgebrochen werden, um die Geburt eines kranken Kindes zu verhindern.
Kann man Kinder bekommen, wenn man die Sichelzellkrankheit hat?
Ja. Wenn jemand die
Sichelzellkrankheit hat und Kinder haben möchte, ist es
allerdings unbedingt notwendig, durch eine Hämoglobin-Analyse
herauszufinden, ob der Partner (die Partnerin) eventuell
Träger der Erkrankung ist. In einem solchen Fall ist die
Wahrscheinlichkeit, daß ein krankes Kind geboren wird, 50%, da
vom Elternteil, das die Sichelzellkrankheit hat, ja nur das
Sichel-Gen vererbt wird. Es sollte deshalb unbedingt in der
Früh-Schwangerschaft die Pränatal-Diagnostik durchgeführt
werden. Ist der Partner eines Sichelzellpatienten kein Träger,
sind alle Kinder dieses Paares Träger, haben also nicht die
Erkrankung.
Frauen mit
Sichelzellkrankheit haben oft in der Schwangerschaft mehr
Schmerzkrisen oder andere Krankheits-Komplikationen.
Paracetamol und Morphin-Präparate sind erlaubt in der ganzen
Schwangerschaft, aber kein Novalgin. Ibuprofen darf nur bis
zur 28. Schwangerschaftswoche genommen werden. Schwangere, die
eine Sichelzellkrankheit haben, müssen in der Schwangerschaft
sehr sorgfältig überwacht werden.
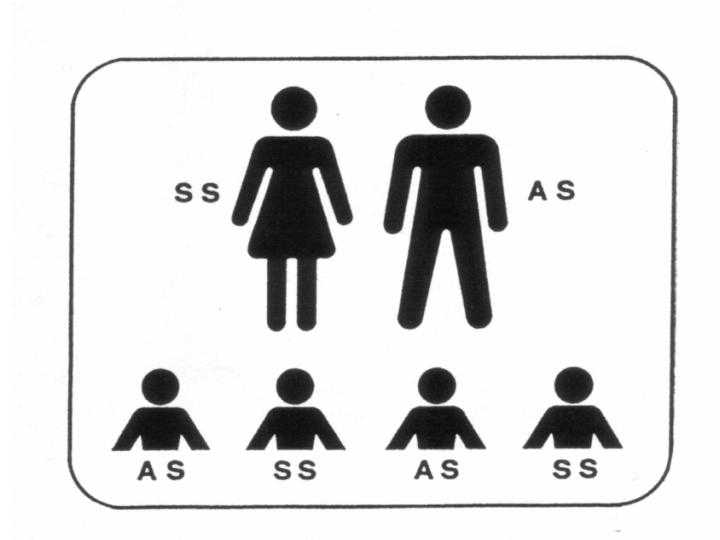
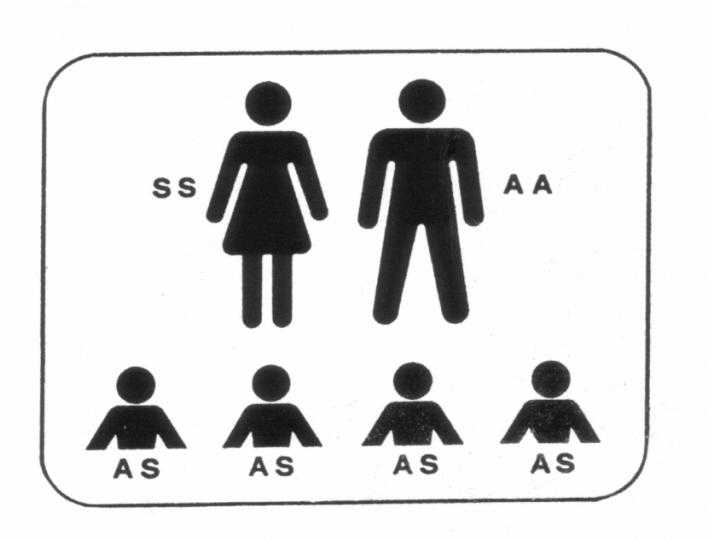
Abb.3
links (oben) :
Sichelzellpatient (SS) mit Partner, der Träger (AS) ist:
Kinder sind krank (SS) oder Träger (AS)
Abb.3 rechts
(unten) : Sichelzellpatient (SS) mit gesundem
Partner (AA): Alle Kinder sind Träger (AS)
Internet Neufassung herausgegeben Frühjahr 2022
Dr.med. Roswitha Dickerhoff
Praxis Prof. Dr. Stefan Eber
Waldfriedhofstr. 73
81377 München
Tel 089 7140975
Fax 089-38366803
E-mail: r.dickerhoff(at)t-online.de
Seit 2008 gibt es in Deutschland eine Selbsthilfegruppe für Sichelzell - und Thalassämiepatienten
Weitere nützliche Verbindungen (auch in anderen Sprachen)
unter:
http://www.ist-ev.org/weiterfuehrende-links